
文 | 千芯科技董事長 陳巍
就在前幾天,迄今為止參數最多、規模最大的蛋白質預測模型ESMFold被Meta官宣了,甚至有研究者宣稱該模型又大又好,足以碾壓Google在2021年推出的AlphaFold2。

▲ESMFold與通訊作者Meta AI的Alexander
這一消息著實讓學術界和工業界震撼,要知道這些大的模型,無論訓練還是使用,都得有妥妥的“鈔能力”,如果模型越來越小,說不定就不需要更大算力的芯片了。(當然事實并非如此)甚至LeCun大牛都發推為ESMFold背書,稱之為“Super-fast and accurate”。
從氨基酸序列預測蛋白質結構是自然科學中長期存在的重大挑戰。在基于進化的算法中,AlphaFold2可以說是目前解決該問題最成功的。它通過在多序列輸入、進化同源物對齊序列和可選結構模板上訓練端到端神經網絡,取得了突破性成就,大大加速了“生命元宇宙”的構建。
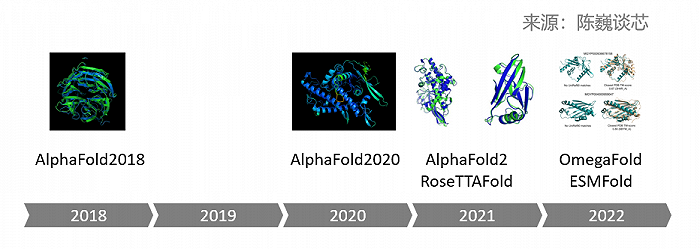
▲蛋白質預測AI大模型的進化
而Meta團隊的ESMFold蛋白質模型只需要一個序列作為輸入,該模型背后的團隊由Meta AI(原Facebook AI)的資深研究科學家Alexander Rives主導。該團隊專注于大規模蛋白質序列和結構數據的無監督表示學習模型研究。Alexander本人同時也是Fate Therapeutics、Syros Pharma、Kallyope的聯合創始人,妥妥的科創家。
那ESMFold真的能碾壓AlphaFold2嗎?讓我們先來回顧下什么是蛋白質結構預測,然后再深入分析ESMFold的網絡結構。
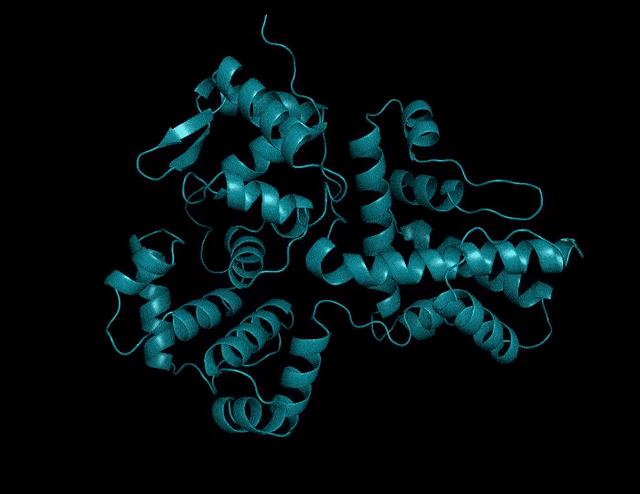
▲ESMFold預測的結構
論文鏈接:https://doi.org/10.1101/2022.07.20.500902
01.什么是蛋白質結構預測?
首先,蛋白質結構是指各種蛋白質分子的空間結構。由線性氨基酸組成的蛋白質,需要折疊(Fold)成特定的空間結構,才具有相應的生理活性和生物學功能。
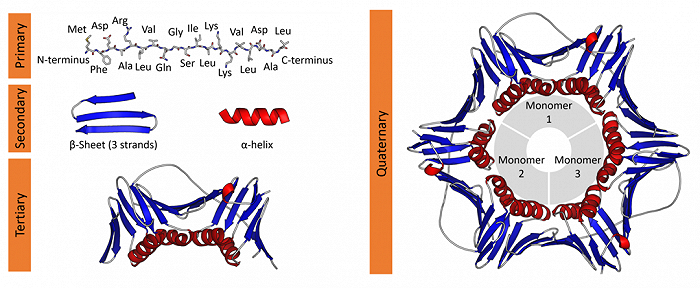
▲蛋白質的四級結構
蛋白質的分子結構可劃分為四級,以描述其不同層級的特征:
蛋白質一級結構:組成蛋白質多肽鏈的線性氨基酸序列。
蛋白質二級結構:依靠不同氨基酸之間的C=O和N-H基團間的氫鍵形成的穩定結構,主要為α螺旋和β折疊。
蛋白質三級結構:通過多個二級結構元素在三維空間的排列所形成的一個蛋白質分子的三維結構。
蛋白質四級結構:用于描述由不同多肽鏈(亞基)間相互作用形成具有功能的蛋白質復合物分子。
我們所說的蛋白質結構預測(Protein Structure Prediction),就是指從蛋白質的氨基酸序列中預測蛋白質的三維結構。也就是說,從蛋白質的一級結構預測其折疊和二級、三級、四級結構。
DeepMind(Google旗下)的AlphaFold2在蛋白質結構預測大賽CASP 14中,對大部分蛋白質結構的預測與真實結構只差一個原子的寬度,達到接近冷凍電鏡等復雜儀器檢測的水平。這一巨大進步被Nature和Science選為2021年度十大科學突破。
根據不同的氨基酸和序列,蛋白質能折疊成的構型數量是一個天文數字,因此很難用常規方法進行蛋白質結構的準確預測。例如,目前實驗的方法(例如冷凍電鏡)至今才能解出10萬的蛋白質結構。
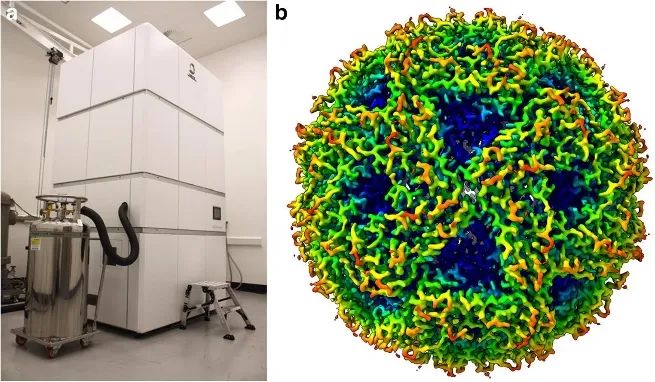
▲冷凍電鏡及其圖像
因此,使用AI的方法,加速對蛋白質結構的解析,分析其組成和功能,就成了生物界和醫藥界的爭相推進的重要工作。
02.ESMFold的“魔幻效果”
ESMFold與AlphaFold2和RoseTTAFold對多序列輸入的蛋白質結構預測具有相當的準確度。但ESMFold突出優勢在于,其計算速度比AlphaFold2快一個數量級,能夠在更有效的時間尺度上探索蛋白質的結構空間。
過去,AlphaFold2和RoseTTAFold在原子分辨率蛋白質結構預測問題上取得了突破性成功,但依賴于使用多序列比對(Multiple Sequence Alignment,簡寫為MSA)和相似蛋白質結構的模板來實現最優表現。
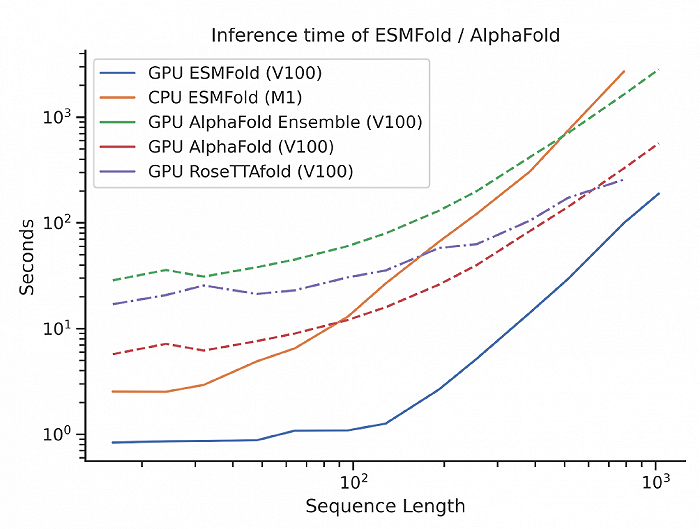
▲ESMFold模型具有比AlphaFold2更高的速度
ESMFold使用ESM-2學習的信息和表示來執行端到端的3D結構預測,特別是僅使用單個序列作為輸入(AlphaFold2需要多序列輸入),方便研究者在使用時通過模型縮放,將模型大小控制在數百萬到數十億量級參數。需要注意的是,隨著模型大小的增加,可觀察到預測準確性的持續提升。(還是“越大越準”)
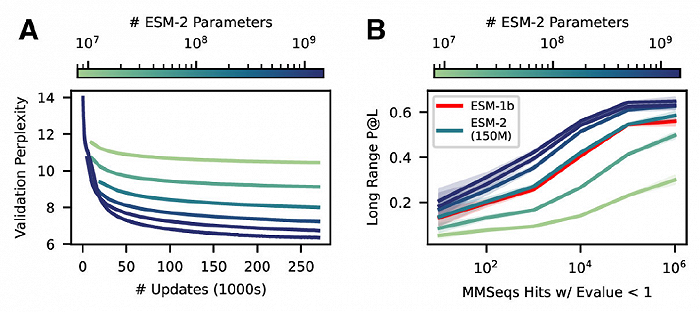
▲ESM-2模型隨著參數量升高精度升高
由于ESMFold的預測速度比現有的其他原子分辨率蛋白質結構預測模型快一個數量級,因此ESMFold可以幫助快速構建蛋白質結構數據庫。使用ESMFold,可以快速計算100萬個預測結構,這些結構代表了蛋白質預測空間的不同子集,其中大多數沒有注釋的結構或功能。
而且ESMFold的大部分高置信度預測與已知的實驗結構的相似度都很低,這表明了通過AI計算獲得的基因組蛋白的結構新穎性。
值得注意的是,許多高置信度結構與UniRef90中的結構也具有低序列相似性,說明該模型具有超出其訓練數據集的泛化能力,實現了基于結構的蛋白質功能預見能力。
據此,研究人員認為,ESMFold可以幫助理解那些超出現有認知的蛋白質結構。
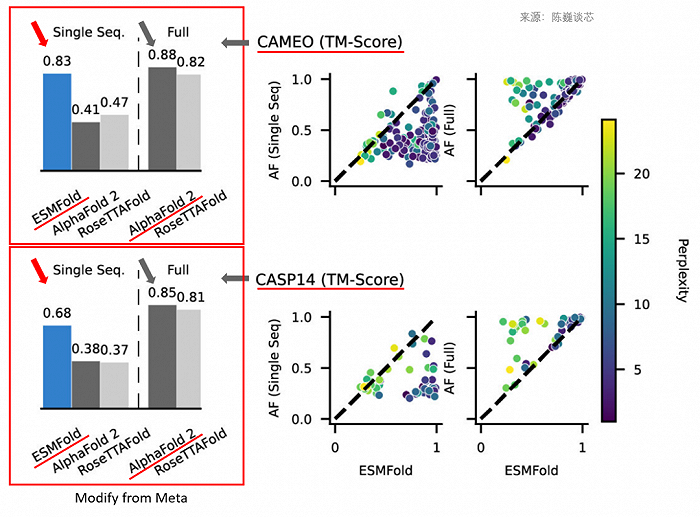
▲ESMFold在單序列輸入時預測精度明顯好于AlphaFold2
雖然ESMFold速度很高,精度也不錯,特別是在單序列輸入的時候精度明顯好于AlphaFold2。但我們也要看到,ESMFold在多序列輸入的情況下,其精度比AlphaFold2還是略有差距。
03.ESMFold網絡結構
與AlphaFold2模型類似,ESMFold模型的架構也可以分為四部分:數據解析部分、編碼器部分(Folding Trunk)、解碼器部分(Structure Module)、循環部分(Recycling)。
ESMFold和AlphaFold2之間的一個關鍵區別是使用語言模型表示來消除對顯式同源序列(以MSA的形式)作為輸入的要求。
語言模型表示作為輸入提供給ESMFold的折疊主干。通過將處理MSA的計算量大的Folding Block模塊替換為處理序列的Tranformer模塊來簡化AlphaFold2中的Evoformer。這種簡化或優化意味著ESMFold會比基于MSA的模型快得多。
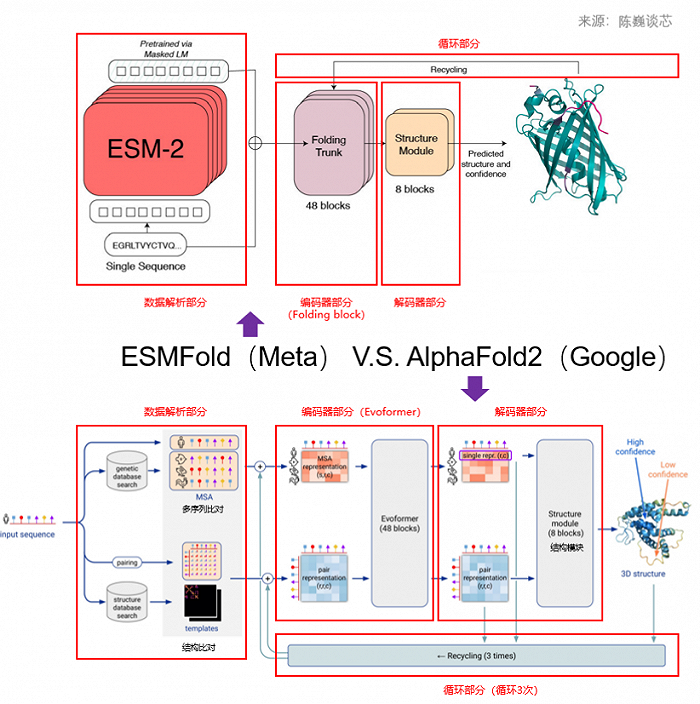
▲ESMFold與AlphaFold2對比
在AlphaFold2和RoseTTAFold中使用MSA和模板會導致兩個瓶頸。
首先,可能需要基于CPU檢索和對齊MSA和模板。這是由于AlphaFold2和RoseTTAFold不是二維序列嵌入狀態,而是使用軸向注意力對應于MSA的三維內部狀態進行操作,即使使用GPU,這一計算的代價也不菲。
相比之下,ESMFold是一個完全端到端的序列結構預測器,可以完全在GPU上運行,無需訪問任何外部數據庫。
例如在單個NVIDIA V100 GPU上,使用較少參數的ESMFold在14.2秒內對具有384個殘基的蛋白質進行預測,可比單個AlphaFold2模型快6倍。而在較短的序列上,我們甚至看到了約60倍的改進。
速度的數量級提高是ESMFold優于AlphaFold2的獨特優勢,使我們能夠在比現有方法更短的時間尺度內構建大量預測結構。考慮到可用序列數據的規模,這一點尤其重要。
例如,AlphaFold2蛋白質結構數據庫的初始版本發布時具有約36萬個預測結構,截至2022年7月則包含約99.5萬個預測,這比目前許多蛋白質序列數據庫小幾個數量級。
04.數據解析部分與解碼器的深度分析
數據解析部分用于輸入序列和數據庫的解析,為編碼器提供輸入。
在AlphaFold2模型中,數據解析部分使用了氨基酸序列數據庫和結構數據庫,分別用于相近序列的比對和結構模板的配對。
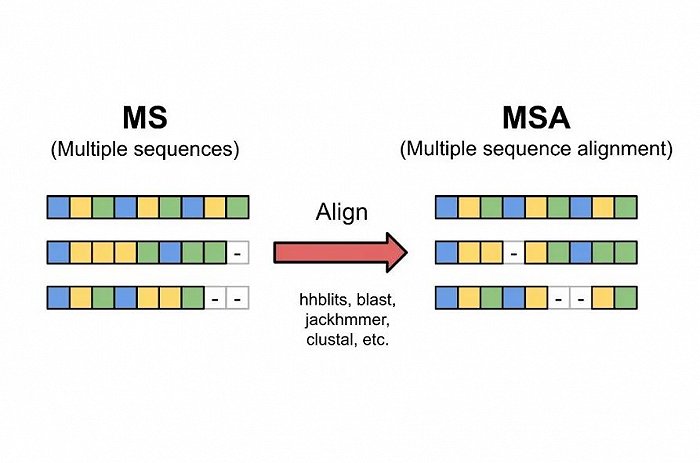
▲AlphaFold2多序列比對示意
生物信息學的基礎是基于這樣的一個假設:序列相似,結構相似,功能相似。一般認為相近的序列或者相近的結構會衍生出相近的功能域。
1)序列數據庫被用于多序列比對(Multiple Sequence Alignment,MSA),即在序列數據庫中檢索與輸入序列接近的數據庫序列。
2)結構數據庫則用于結構匹配,尋找與輸入序列的結構接近的已知結構模板。
然后序列比對與結構比對的結果作為輸入傳輸給編碼器部分。
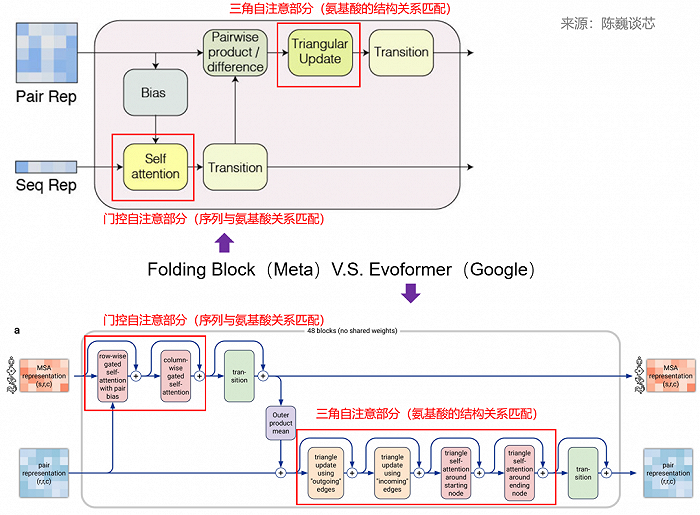
▲ESMFold Folding Block與AlphaFold2 Evoformer結構對比
解碼器部分即Folding Trunk,一共48層。
ESMFold與AlphaFold2的一個關鍵區別是,ESMFold使用語言模型表示,消除了對明確的同源序列(以MSA的形式)作為輸入的需要。
ESMFold通過用一個處理序列的Transformer模塊取代處理MSA的計算昂貴的網絡模塊,簡化了AlphaFold2中的Evoformer。這種簡化意味著ESMFold的速度大大提高,遠高于基于MSA的模型。
05.結語
作為蛋白質結構預測大模型,ESMFold獲得準確原子分辨率結構預測的推斷(Inferenc)速度比AlphaFold2提高了約一個數量級。特別是在實際計算中,這一速度優勢表現的更加明顯。這是由于ESMFold削減了搜索多序列來構建MSA的計算量。
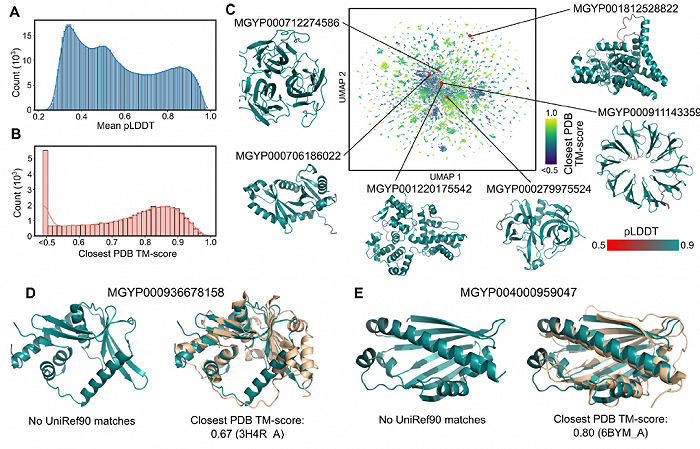
▲ESMFold用于探索宏基因組結構空間
推斷速度優勢使得基于計算有效映射大型宏基因組序列數據庫的結構空間成為可能。
除了用于識別遠同源性外,ESMFold還可以被用于進行快速準確的結構預測,并在實際時間尺度內獲得數百萬個預測結構,進一步幫助發現新的蛋白質結構和功能。這相當于在使用AI計算來構建生命的“元宇宙”。
150億參數大模型,10x倍速度提升。雖然Meta ESMFold精度上沒能做到全面“碾壓”AlphaFold2,但畢竟“唯快不破”,對于蛋白質結構解析與預測、構建大型宏基因組結構數據庫有著巨大的推動作用。
參考文獻:
Zeming Lin et. al., Language models of protein sequences at the scale of evolution enable accurate structure prediction, https://www.biorxiv.org/content/10.1101/2022.07.20.500902v1
Jumper, J. et al., Highly accurate protein structure prediction with AlphaFold, Nature (2021):1-11.